在过去的日子里——真正的过去——设计材料的任务是繁重的。在超过1000年的时间里,研究人员试图通过将铅、汞和硫等物质以他们希望的恰当比例混合来制造黄金。甚至像第谷·布拉赫、罗伯特·波义耳和艾萨克·牛顿这样的著名科学家也曾尝试过我们称之为炼金术的无果之举。
当然,材料科学已经取得了长足的进步。在过去的150年里,研究人员得益于元素周期表,这告诉他们不同的元素具有不同的性质,且一个元素不能神奇地转变为另一个。此外,在过去十年左右,机器学习工具显著提升了我们确定各种分子和物质的结构及物理性质的能力。由麻省理工学院核工程东京电力公司教授及材料科学与工程教授李居明领导的一组新研究,承诺在促进材料设计方面实现重大能力飞跃。他们的研究结果已在2024年12月的自然计算科学期刊中报告。
目前,大多数用于表征分子系统的机器学习模型基于密度泛函理论(DFT),该理论通过观察电子密度分布——基本上是位于分子附近每个给定点周围单位体积内的平均电子数——提供了一种量子力学方法来确定分子或晶体的总能量。(沃尔特·科恩在60年前共同发明了这一理论,并因此在1998年获得诺贝尔化学奖。)尽管该方法非常成功,但李指出它也有一些缺点:“首先,准确性并不均匀出色。其次,它只告诉你一件事:分子系统的最低总能量。”
“伴侣疗法”来救援
他的团队现在依赖于一种不同的计算化学技术,也源于量子力学,称为耦合簇理论(CCSD(T))。李评论道:“这是量子化学的金标准。”CCSD(T)计算的结果比DFT计算的结果更为准确,并且可以与目前从实验中获得的结果一样可靠。问题在于,在计算机上进行这些计算非常缓慢,他说,“而且扩展性很差:如果你将系统中的电子数量加倍,计算成本将增加100倍。”因此,CCSD(T)计算通常仅限于原子数量较少的分子——大约10个原子。超过这个数量的分子计算将耗时太长。
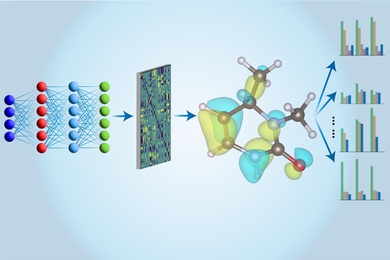
这就是机器学习发挥作用的地方。首先在常规计算机上进行CCSD(T)计算,然后将结果用于训练李和他的同事们特别设计的新型架构的神经网络。经过训练后,神经网络可以利用近似技术更快地执行这些相同的计算。此外,他们的神经网络模型可以提取关于分子更多的信息,而不仅仅是其能量。“在之前的工作中,人们使用多种不同的模型来评估不同的性质,”麻省理工学院材料科学与工程博士生唐浩说。“在这里,我们只使用一个模型来评估所有这些性质,这就是我们称之为‘多任务’方法的原因。”
“多任务电子哈密顿网络”(MEHnet)揭示了许多电子性质,如偶极矩和四极矩、电子极化率以及光学激发间隙——将电子从基态转移到最低激发态所需的能量。“激发间隙影响材料的光学性质,”唐解释道,“因为它决定了分子可以吸收的光的频率。”他们的CCSD训练模型的另一个优点是,它可以揭示不仅是基态的性质,还有激发态的性质。该模型还可以预测与分子振动性质相关的红外吸收光谱,其中分子内原子的振动相互耦合,导致各种集体行为。
他们的方法的强大之处在于网络架构。唐表示,借鉴麻省理工学院助理教授特斯·斯米特的工作,团队利用了一种所谓的E(3)-等变图神经网络,“其中节点代表原子,连接节点的边代表原子之间的键。我们还使用了定制的算法,将与人们在量子力学中计算分子性质相关的物理原理直接纳入我们的模型。”
测试,1,2,3
在对已知烃分子的分析中,李等人的模型表现优于DFT模型,并与从已发表文献中获得的实验结果密切匹配。
北卡罗来纳大学夏洛特分校的材料发现专家朱强(未参与本研究)对迄今为止取得的成就印象深刻。“他们的方法能够在小数据集上进行有效训练,同时在准确性和计算效率上优于现有模型,”他说。“这项令人兴奋的工作展示了计算化学与深度学习之间强大的协同作用,为开发更准确和可扩展的电子结构方法提供了新思路。”
麻省理工学院的团队首先将他们的模型应用于小型非金属元素——氢、碳、氮、氧和氟,这些元素可以用来制造有机化合物——并随后转向研究更重的元素:硅、磷、硫、氯,甚至铂。在对小分子进行训练后,该模型可以推广到越来越大的分子。“之前,大多数计算仅限于使用DFT分析数百个原子,而使用CCSD(T)计算仅限于数十个原子,”李说。“现在我们谈论的是处理数千个原子,最终可能是数万个。”
目前,研究人员仍在评估已知分子,但该模型可以用于表征尚未见过的分子,以及预测由不同种类分子组成的假想材料的性质。“我们的想法是利用我们的理论工具挑选出满足特定标准的有前景候选者,然后建议实验人员进行检查,”唐说。
一切都与应用有关
展望未来,朱对可能的应用持乐观态度。“这种方法具有高通量分子筛选的潜力,”他说。“这是一个在识别具有理想性质的新分子和材料时,化学准确性至关重要的任务。”
一旦他们展示出分析可能有数万个原子的巨大分子的能力,李表示,“我们应该能够发明新的聚合物或材料”,这些材料可能用于药物设计或半导体设备。对更重的过渡金属元素的研究可能会导致新材料的出现,尤其是在电池领域——这是一个急需的领域。
李所看到的未来是广阔的。“这不再仅仅是一个领域,”他说。“我们的最终目标是以低于DFT的计算成本覆盖整个元素周期表,并达到CCSD(T)级别的准确性。这应该使我们能够解决化学、生物学和材料科学中的广泛问题。目前很难知道,这个范围可能有多广。”
这项工作得到了本田研究所的支持。唐浩感谢Mathworks工程奖学金的支持。本研究中的计算部分是在Matlantis高速通用原子模拟器、德克萨斯先进计算中心、麻省理工学院超级云和国家能源研究科学计算中心进行的。



